Etimologia
A Chicungunha é um aportuguesamento de Chikungunya, o nome da doença na língua Kimaconde, um dos idiomas oficiais da Tanzânia e significa “inclinou-se ou contorceu-se de dor”, onde foi documentada a primeira epidemia da doença em 1953. O termo provém da raiz verbal kungunyala, e significa "tornar-se dobrado ou contorcido" (referindo-se à aparência dos pacientes que se inclinam por causa da dor nas articulações que a doença provoca), em referência à aparência curvada dos pacientes, motivada pelas intensas dores articulares e musculares, características da doença. Em Angola (África) a doença é popularmente conhecida por Catolotolo, palavra proveniente do Quimbundo Katolotolu, derivação do verbo Kutolojoka ("ficar alquebrado").
Hoje, a Chicungunha já foi identificada em mais de 60 países na Ásia, África, Europa e nas Américas, e ocorre na África, Ásia e no subcontinente indiano. Nas últimas décadas, os mosquitos vetores da Chicungunha se espalharam pela Europa e pelas Américas. Em 2007, a transmissão da doença foi relatada pela primeira vez durante um surto no noroeste da Itália. Desde então, surtos também foram registrados na França e na Croácia.
Nos continentes americanos, o número de casos reportados até 2015 era de 37.480 e o de suspeitas era de 693.489, sendo 356.079 na Colômbia.
Segundo a Organização Pan-Americana da Saúde, a letalidade da Chicungunha é rara, sendo menos frequente que nos casos de Dengue.
Por outro lado, a doença gera um grande impacto social por causa do alto número de casos; da incapacidade de trabalhar, às vezes, a longo prazo; das consequências dos efeitos colaterais de medicamentos inapropriados e das consequências da não obtenção de um diagnóstico preciso.
No Brasil, casos da doença foram detectados pela primeira vez em agosto de 2010.
Causa
O vírus Chicungunha é um RNAss - Ácido Ribonucleico de Hebra Simple Vírus, do gênero Togaviridae, esférico, com envelope, arbovírus transmitido por mosquitos Aedes, atualmente encontrado em todos os continentes.
Transmissão
A transmissão do vírus Chicungunha (CHIKV) é feita através da picada de insetos-vetores do gênero Aedes, que em cidades é principalmente pelo Aedes Aegypti e em ambientes rurais ou selvagens pode ser por Aedes Albopictus. Embora a transmissão direta entre humanos não esteja demonstrada, há de se considerar a possibilidade da transmissão in utero da mãe para o feto.
O período de incubação do vírus é de 4 a 7 dias, e a doença, na maioria dos casos, é auto-limitante. A mortalidade em menores de um ano é de 0,4%, podendo ser mais elevada em indivíduos com patologias associadas.
Epidemiologia
Casos da febre Chicungunha foram relatados na Tailândia (em 1953), Indonésia, Taiwan, Singapura, Malásia, Sri Lanka, ilhas Maldivas, Quênia (em 2004), Comores (em 2005), Mayotte, Seychelles, Maurícia, Reunião (2005-2006) e Índia (2006), e, em menor intensidade, na Itália, Martinica, Guadalupe, Guiana Francesa, Estados Unidos e Brasil (em 2010).
Um surto de Chicungunha foi relatado em 2006 em Andra Pradexe (Índia), mesma época em que casos alóctones foram relatados em diversos países europeus.
Chegou nas Américas apenas em 2014 e segue expandindo causando uma pandemia. Desde então mais de 1,7 milhões de casos suspeitos notificados à Organização Pan-Americana da Saúde. O diagnóstico é difícil pois é muito similar a outras viroses. Causou surtos recentes (2010-2015) por todas as Américas, Europa, África, China, sudeste Asiático e ilhas do pacífico.
A existência de grandes cidades densamente povoadas onde existam os insetos vetores da doença, bem como o aumento do número de viagens entre países e intercontinentais facilitam a disseminação do vírus.
Brasil
Os primeiros casos confirmados no Brasil, em 2010, referem-se a dois pacientes do sexo masculino (de 41 e 55 anos, em São Paulo) que apresentaram os sintomas depois de uma viagem à Indonésia. A terceira paciente, uma paulista de 25 anos, esteve na Índia.
Em junho de 2014 foram confirmados seis casos no Brasil de soldados que retornaram de uma missão no Haiti. Segundo dados publicados pelo Ministério da Saúde, porém, no dia 15 de outubro de 2014, foram confirmados 337 casos no país, sendo 274 apenas na cidade de Feira de Santana, na Bahia. Em 2015 ocorreu um surto na América do sul nos primeiros quatro meses deste ano com estimativa de 10 mil casos e 113 mortes. Estima-se que 2.500 desses casos foram no Brasil, a maioria dos casos na Bahia, Minas Gerais e São Paulo. E até agora em 2016 se encontra casos extremos.
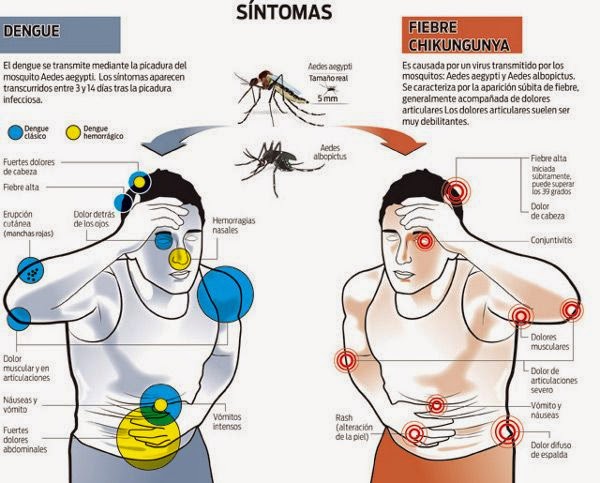
Sinais e sintomas
Os sintomas da febre Chicungunha são característicos de uma virose, e portanto, inespecíficos. Os sintomas iniciais são febre acima de 39º C, de início repentino, dores intensas nas articulações de pés e mãos, dedos, tornozelos e pulsos, dores de cabeça, dores musculares e manchas vermelhas na pele. O diagnóstico diferencial com a febre hemorrágica da dengue é extremamente importante, razão pela qual, ao aparecimento dos sintomas é fundamental buscar socorro médico.
Diferentemente da dengue, doença viral transmitida pelos mesmos mosquitos vetores, uma parte dos indivíduos infectados pode desenvolver a forma crônica da doença, com a permanência dos sintomas, que podem durar entre 6 meses e 1 ano.



















{kind=link}